Ruxolitinib for Treating Steroid-Refractory Acute Graft-Versus-Host Disease in an Infant with Malignant Osteopetrosis Who Received Double-Unit Umbilical Cord Blood Transplantation
Article information

Abstract
A 3-month-old male infant was transferred to our hospital due to bicytopenia. His bone marrow biopsy showed irregular bony trabeculae with cartilaginous core, which was consistent with osteopetrosis. In the genetic test, c.242del (p.Pro81Argfs*85) in TCIRG1 was found to be homozygotic, thus he was diagnosed with malignant infantile osteopetrosis. At 6 months of age, he received double-unit umbilical cord blood transplantation (UCBT) with the conditioning regimen including busulfan, cyclophosphamide, and rabbit anti-thymocyte globulin. Initially, single UCB was infused to the patient, but the post infusion viability of the UCB was unexpectedly low. Thus, another UCB was additionally infused. Cyclosporine and mycophenolate mofetil were used for graft-versus-host disease (GVHD) prophylaxis. Neutrophils and platelets were engrafted on day +13 and +33, respectively. With engraftment, he showed overall grade 4 acute GVHD involving the skin and gut, which was refractory to corticosteroids. Despite treating with low-dose weekly methotrexate (10 mg/m2) and oral beclomethasone, his symptoms persisted. After treating with ruxolitinib 2.5 mg/day for 2 weeks, and 5 mg/day thereafter, his diarrhea stopped in 2 weeks and his skin symptoms gradually improved over 3 months. The short tandem repeats showed 100% donor chimerism at 1 and 3 months after UCBT. Currently, 4 months after UCBT, he is 10 months old. The oral prednisolone has been tapered to 0.6 mg/kg/day, and the dose of ruxolitinib was decreased to 2.5 mg/day without recurrence of GVHD. We plan to taper off the immunosuppressive agents if his GVHD symptoms do not recur.
Introduction
Osteopetrosis includes a group of heterogeneous genetic disorders characterized by dysfunctional osteoclasts that result in an inappropriate bone marrow (BM) cavity for hematopoiesis and extramedullary hematopoiesis [1]. There may be three types of osteopetrosis based on inheritance, namely autosomal dominant, autosomal recessive (AR), and X-linked [2]. The AR osteopetrosis has an incidence of 1:250,000 births, with high degree of parental consanguinity [2]. The AR osteopetrosis is also called ‘malignant infantile’, since it is diagnosed very early after birth and can be lethal [3]. If left untreated, the probability of survival of individuals with AR osteopetrosis until the age of 6 years is only about 30% due to BM failure [4]. Since osteoclasts are hematopoietically derived, osteopetrosis can be treated by allogenic hematopoietic stem cell transplantation [5]. This study aimed to report an infant with malignant osteopetrosis, who received double-unit umbilical cord blood transplantation (UCBT). He developed steroid-refractory acute graft-versus-host disease (GVHD) thereafter, which was subsequently managed with ruxolitinib.
Case
A 3-month-old male infant was transferred to our hospital due to laboratory abnormalities. He was born as late preterm infant at 35 weeks by normal vaginal delivery. His birth weight was 2.68 kg, which was appropriate for gestational age. However, at the time of visiting our hospital, he had failure to thrive; his height was 50.7 cm (< 3 percentile) and weight 4.72 kg (< 3 percentile). Upon physical examination, he was found to have hepatosplenomegaly. His initial laboratory tests showed the following: white blood cell count 16.99 × 109/L (reference, 4–10 × 109/L), hemoglobin 9.2 g/dL (reference, 13.0–17.0 g/dL), platelet count 75 × 109/L (130–400 × 109/L), reticulocyte 11.5% (reference, 0.5–1.5%), total bilirubin 1.34 mg/dL (0.2–1.2 mg/dL), direct bilirubin 0.47 mg/dL (reference, 0–0.42 mg/dL), ALP 1,072 U/L (reference, 40–129 U/L), and LDH 2,364 U/L (reference, 270–450 U/L).
His BM biopsy showed irregular bony trabeculae with cartilaginous core, which was consistent with osteopetrosis (Fig. 1). Besides, the laboratory findings were also compatible with osteopetrosis—extramedullary hematopoiesis and hemolytic anemia associated with splenomegaly. Skeletal survey of the skull and extremities revealed diffused dense skeletal system with loss of cortico-medullary differentiation and irregular lucency involving the entire metaphyses of the long bones, which were consistent with osteopetrosis (Fig. 2). The results of the genetic analysis revealed that the known pathogenic variant, c.242del (p.Pro81Argfs*85) in TCIRG1, was homozygotic. Thus, the patient was diagnosed with osteopetrosis, autosomal recessive 1 (OMIM #259700).

Bone marrow biopsy specimen of an infant with malignant osteopetrosis. The stained slide showed irregular bony trabeculae with cartilaginous core, which was consistent with osteopetrosis (H&E, 40×).
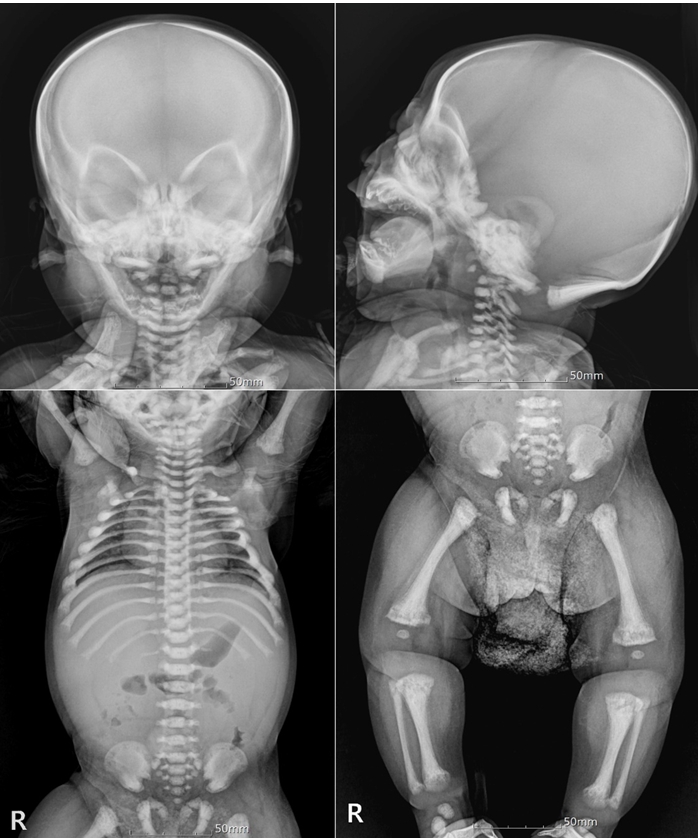
The radiological findings of an infant with osteopetrosis. Skeletal survey of the skull and extremities revealed diffused dense skeletal system, loss of cortico-medullary differentiation, ‘bone with bone appearance’, irregular lucency involving the entire metaphyses of the long bones, all consistent with osteopetrosis. Chest x-ray showed diffusely increased density of axial skeleton and expansion of the anterior rib ends at the costochondral junctions.
At 6 months of age, he received double-unit UCBT, since he had no sibling, and there was no matched or mismatched unrelated donor. The conditioning regimen was as follows: intravenous busulfan 4.0 mg/kg (days −8 to −5), cyclophosphamide 60 mg/kg (days −3, −2), and rabbit anti-thymocyte globulin 2.5 mg/kg (days −3 to −1). Defibrotide was infused for prophylaxis of veno-occlusive disease as 6.25 mg/kg/dose q 6rs. Cyclosporine and mycophenolate mofetil were used for GVHD prophylaxis. Initially, single UCB with 3 loci mismatch was infused to the patient; however, the post-infusion viability of UCB was unexpectedly low at 13% (TNC 2.9 × 107/kg and CD34 0.07 × 105/kg). Thus, another UCB with 3 loci and 1 allele mismatch, prepared as a candidate, was additionally infused (TNC 11.5 × 107/kg and CD34 4.3 × 105/kg). He developed engraftment syndrome, which was subsequently managed with corticosteroid and ventilator care. Neutrophils and platelets were engrafted on days +13 and +33, respectively.
With engraftment, he showed overall grade 4 acute GVHD involving the skin (stage 4) and gut (stage 3), which was refractory to 2 mg/kg of methylprednisolone. The skin and large intestine were biopsied and the samples were sent to the pathology department. The skin specimen showed mild spongiosis, intraepidermal lymphocytic infiltrate, and scattered dyskeratotic/apoptotic keratinocytes (Fig. 3A). The large intestinal mucosa revealed mild lymphocytic infiltrate in the lamina propria and prominent apoptotic bodies in the crypts (Fig. 3B). Despite the addition of high dose corticosteroid (10 mg/kg/day for 3 days), low-dose weekly methotrexate (10 mg/m2), and oral beclomethasone, his symptoms persisted without any clinical improvement.

Biopsy specimen in a patient with steroid-refractory grade IV acute graft-versus-host disease. (A) The skin specimen showed mild spongiosis, intraepidermal lymphocytic infiltrate, and scattered dyskeratotic/apoptotic keratinocytes (H&E, 100×). (B) The large intestinal mucosa revealed mild lymphocytic infiltrate in the lamina propria and scattered intraglandular apoptotic bodies (H&E, 100×).
In Korea, administration of ruxolitinib for the treatment of GVHD in children is not yet permitted. Therefore, this study was approved by the Institutional Review Board of Keimyung University Dongsan Hospital (Approval No. 2020-09-016). Informed consent was obtained from the patient’s parents. Considering his age (7 months) and weight (6.8 kg), we initially prescribed oral ruxolitinib 2.5 mg/day (1.25 mg/dose q 12 hrs) for 2 weeks, and then increased the dose of ruxolitinib as 5 mg/day (2.5 mg/dose q 12 hrs) as powder. His diarrhea stopped 2 weeks after initiation of ruxolitinib. His skin symptoms gradually improved over 3 months after ruxolitinib administration. The change in his skin before and after 4 weeks and 12 weeks of ruxolitinib is shown in Fig. 4. He additionally received fluconazole for antifungal prophylaxis, cotrimoxazole against Pneumocystis jirovecii, and viral prophylaxis with acyclovir. There was no adverse event including cytopenia or infection during ruxolitinib treatment for acute GVHD. The short tandem repeats showed 100% donor chimerism at 1 and 3 months after UCBT. Currently, he is 10 months old and at 4 months post UCBT. The oral prednisolone has been tapered to 0.6 mg/kg/day, and the dose of ruxolitinib was decreased to 2.5 mg/day without recurrence of GVHD. We plan to taper off the immunosuppressive agents if his GVHD symptoms do not recur.
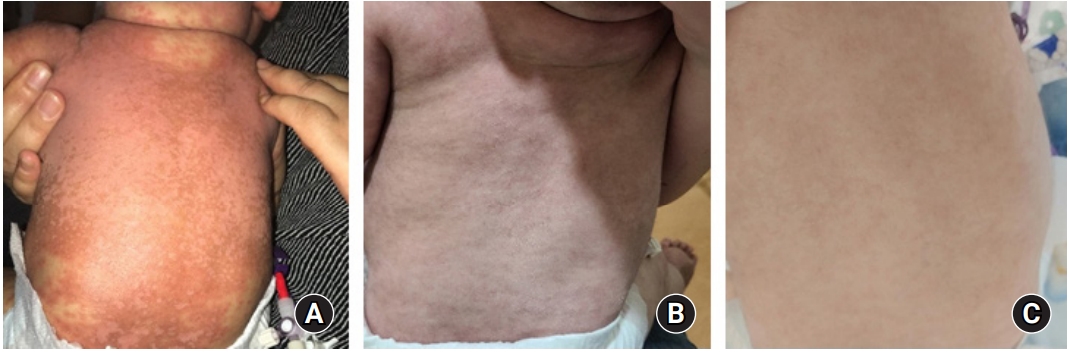
Progression of skin manifestations in an infant with malignant osteopetrosis after treatment of steroid-refractory acute graft-versus-host disease with ruxolitinib. (A) Before ruxolitinib. (B) 4 weeks after ruxolitinib. (C) 12 weeks after ruxolitinib.
Discussion
The patient presented here had the known pathogenic variant of AR osteopetrosis, c.242del (p.Pro81Argfs*85) in TCIRG1, which was homozygotic. Since TCIRG1 encodes V-type proton ATPase, its mutations result in defects in the proton-pumping function of V-ATPase and vesicle trafficking or fusion of osteoclasts [2]. The mutation in TCIRG1 most typically accompanies approximately half the cases of AR osteopetrosis [2,3]. The patient presented here had the typical phenotype of malignant infantile osteopetrosis with AR inheritance, with ineffective hematopoiesis from BM and extramedullary hematopoiesis in early infancy. Since he had no sibling and there was no matched or mismatched unrelated donor, he received UCBT.
An alternative donor, UCB, has been widely used for allogenic hematopoietic cell transplantation with a high allowance of HLA disparities [6]. The incidence of GVHD in UCBT is known to be lower than expected given the degree of HLA disparity [6]. It may be associated with functional immaturity of the infused lymphocytes and increased regulatory T cells of UCB [7]. Further, UCB not only has fewer T cells than other donors, but also has T cells with naive phenotype with atypical functional properties and low cytotoxicity [8]. Despite these characteristics of UCB, the present infant patient with osteopetrosis suffered severe acute GVHD, which was refractory to corticosteroid therapy. We speculated that he developed severe acute GVHD due to the unexpected double-unit UCBT and high cell dose.
Since this patient was an infant with low body weight, the cell count of single UCB was initially considered sufficient. However, unexpectedly, viability test after thawing the first UCB showed remarkably decreased viability (13%), which eventually led to the infusion of another UCB. We also considered graft rejection in him under non-malignant condition, i.e. osteopetrosis. The cumulative incidence of grade II–IV acute GVHD is higher among double-unit UCBT recipients than among single-unit UCBT recipients [9,10]. However, transplantation-related mortality was significantly lower after double-unit UCBT, even if the recipients had grade II–IV acute GVHD [9]. The higher cell dose with TNC > 2.5 × 107/kg and CD34 > 1.0 × 105/kg was also associated with the development of grade II–IV GVHD [11]. Acute GVHD is a major cause of morbidity and mortality after allogenic stem cell transplantation, occurring in 30–50% of recipients [12,13]. Although corticosteroid is a standard first-line treatment for acute GVHD, approximately half of the patients show refractoriness to the systemic corticosteroid therapy with high mortality rate [14].
Ruxolitinib is a selective inhibitor of janus kinase (JAK), particularly for the subtypes JAK1 and JAK28 approved for the treatment of myelofibrosis [13]. The JAK signaling pathway also plays an important role in immune cell activation and tissue inflammation in acute GVHD [15]. Ruxolitinib (10 mg twice daily) showed significant improvements in efficacy in a multicenter, randomized, open-label, phase 3 trial in patients ≥ 12 years with steroid refractory acute GVHD after allogenic stem cell transplantation [15]. Salvage treatment with ruxolitinib for steroid refractory GVHD in children has been reported as a retrospective study abroad [16-18]. The pediatric dosing of ruxolitinib for GVHD is not established yet. Based on studies on children abroad, the median dose of ruxolitinib was 12.6 mg/m2/day (range, 6.3–28.7 mg/m2/day) for the children < 6 years old [16]. Approximately, ruxolitinib was administered as 2.5 mg/dose q 12 hours in children < 25 kg, and 5 mg/dose q 12 hours in children ≥ 25 kg [19]. In case of infants abroad, a lower dose of ruxolitinib, 2.5 mg/day was administered for managing acute GVHD [19]. In case of our infant patient, we initially prescribed ruxolitinib as 2.5 mg/day, and then the dose was increased as 5 mg/day because his skin GVHD persisted.
There had been no report about ruxolitinib administration in infants in Korea yet. Further, this is the first report of using ruxolitinib in an infant with osteopetrosis who received double-unit UCBT. The presented infant had a rare disease, namely malignant osteopetrosis, and suffered severe steroid-refractory acute GVHD because of unexpected double-unit UCBT. Nevertheless, it was well managed with ruxolitinib in a very rare and complex situation.
Notes
Conflict of interest
All authors declare no conflicts-of-interest related to this article.