Introduction
Neuroinflammation has exhibited its pivotal role in the development of many neurological illnesses, including Alzheimer’s disease (AD), in response to numerous pathological conditions [1]. In central nervous system (CNS), neuroinflammation is marked as the activation of microglia, high production of pro-inflammatory cytokines, recruitment of T-cells, and neural tissue (cell) damage [2]. Accumulating evidence further indicates that various extrinsic and intrinsic stimuli, such as lipopolysaccharide (LPS), particulate matter (PM), interferon-gamma (IFN-γ), or tumor necrosis factor-alpha (TNF-α), can induce the microglia activation, which results in the abnormal expression of many neuroinflammatory mediators, including inducible nitric oxide synthase (iNOS), TNF-α, and interleukin-1 (IL-1), and IL-6 [3,4].
iNOS is an inflammatory enzyme responsible for the production of nitric oxide (NO) in the human body, including brain [5-7]. We and others have previously reported that LPS induces high expression of iNOS in BV2 cells, a mouse microglial cell line, and the LPS-induced iNOS expression in these cells is mediated through the activation of not only nuclear factor-kappa B (NF-κB), a transcription factor, but also multiple protein kinases, including extracellular signal-regulated kinase (ERK-1/2), p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK-1/2), and protein kinase B [8-10]. Of importance, there is a large body of evidence that overexpression of iNOS (and the resultant NO overgeneration) plays a crucial role in neuroinflammation and neuronal cell death in the damaged CNS [11]. Thus, iNOS and its inhibitor are considered a key molecular target and targeted therapy against neuroinflammatory pathologies [12,13]. It is therefore conceivable that any substance that inhibits iNOS overexpression in the activated microglia or glial cells may be applied as a potential anti-neuroinflammatory agent or drug candidate molecule.
There are no specific drugs currently available to repair the damaged neurons, to induce neurodegeneration, and to prevent neuroinflammation, and current drugs are effective only for reducing the severity of symptoms by limiting the extent of neuroinflammation. A wealth of information exists that non-steroidal anti-inflammatory drugs (NSAIDs), due to their neuroprotective, anti-inflammatory, and antioxidant properties, may be used for preventing and protecting neurodegenerative illnesses [14,15], although they are associated with an increased risk of adverse gastrointestinal, renal and cardiovascular effects. It is documented that their main action mechanisms are the inactivation of cyclooxygenase-2 (COX-2), the inducible enzyme responsible for overproduction of prostaglandins and thromboxanes, known inflammation and pain inducing hormones [16,17] and the inhibition of NF-κB, the transcription factor involved in the transcriptional up-regulation of many inflammatory genes, which include iNOS, COX-2, and cytokines [18].
Emerging study is needed to develop new therapeutic agent to prevent neurological pathogenesis involving neuroinflammation or to slow its progression in the damaged CNS. It is also currently thought that neurodegenerative disorders are considered as a multifactorial disease; hence, the therapeutic strategies to combat neurological pathogenesis and/or neurodegenerative disorders should involve multiple targets instead of a single target or targeted therapy [19-21]. Naproxen and ibuprofen are known NSAIDs with neuroprotective and anti-inflammatory effects. To evaluate the structural requirement and necessity of carboxylic function in the drugs as a part of our ongoing effort to generate multi-target ligands for neurodegenerative diseases, in this study, we newly synthesized a small series of naproxen and ibuprofen amide dimers by the structural modification and tested whether these amide dimers can modulate the LPS-induced iNOS expression in BV2 cells.
Materials and Methods
1. Materials
Primary antibodies for phosphorylated (p-) ERK-1/2, total (T-) ERK-1/2, p-JNK-1/2, T-JNK1/2, p-p38 MAPK, and T-p38 MAPK were purchased from Cell Signaling Tech. (Danvers, Massachusetts, USA). iNOS and IκB-α antibodies were purchased from Santa Cruz biotechnology (Dallas, Texas, USA). Enhanced chemiluminescence (ECL) reagents were purchased from Advansta (San Jose, CA, USA). The anti-actin antibody was obtained from Sigma-Aldrich (St. Louis, Mo, USA).
2. Synthesis of NSAID amide dimers
All chemicals were purchased from commercial sources and used without further purification. Silica gel 60 F-254 (0.25-mm thickness) plates were used for thin-layer chromatography (TLC) analysis and ultraviolet light and/or iodine vapor was for visualization on TLC plates. Silica gel column chromatography used for purification was performed on Merck silica gel 60 (230-400 mesh). The NMR spectra were recorded using a Burker (Fourier 300) 300 MHz or Burker (AVANCE Neo 400) 400 MHz spectrometer. All 1H NMR spectra for samples are reported in δ units (ppm) and are referenced to the peak for tetramethylsilane if conducted in CDCl3, or to the central line of the quintet at 2.49 ppm in d6-DMSO. Spectral data are reported as follows: chemical shift (ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad), coupling constant, and integration value. Coupling constants (J) are reported in hertz.
(2S,2'S)-2,2'-(((2-Hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2-diyl))bis(N-hexylpropanamide) (CNU 015). A mixture of (2S,2'S)-2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2-diyl))dipropionic acid (1, 70 mg, 0.143 mmol), HATU (120 mg, 0.32 mmol), n-hexylamine (31.4 mg, 0.31 mmol) and triethylamine (94 mg, 0.93 mmol) in DMF was stirred at RT. After 16h, the mixture was poured into EtOAc, washed sequentially with diluted HCl, sat. NaHCO3, brine, and dried with Na2SO4. Filtration, evaporation and purification of the residue by column chromatography (silica gel, 33% of EtOAc in hexane with drops of acetic acid) provided compound CNU 015 (88.4 mg, 96.5%) as an slightly brown solid. 1H-NMR (300 MHz, DMSO-d6): δ 7.94 (br t, J = 6.0, 2 H), 7.79-7.70 (m, 6 H), 7.44-7.41 (m, 2 H), 7.33-7.32 (m, 2 H), 7.20-7.17 (m, 2 H), 5.51 (br d, J = 6.0, 1 H), 4.33-4.14 (m, 5 H), 3.70 (q, J = 7.0, 2 H), 3.01 (q, J = 6.0, 4 H), 1.39 (d, J = 9.0, 6 H, overlap with m), 1.34-1.11 (m, 16 H, overlap with d), 0.79 (t, J = 6.0, 6 H).
Di-tert-butyl 2,2'-(((2S,2'S)-2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2-diyl))bis(propanoyl))bis(azanediyl))diacetate (CNU 016). Following the procedure used to prepare CNU 015, using (2S,2'S)-2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2-diyl))dipropionic acid (1, 70 mg, 0.143 mmol), HATU (117.9 mg, 0.31 mmol), glycine 1,1-dimethylethyl ester (tert-Butyl 2-aminoacetate) (42.4 mg, 0.31 mmol) and triethylamine (94 mg, 0.93 mmol) and eluting with a 20% EtOAc in hexane provided CNU 016 (83.6 mg, 82% yield) as an oil. 1H-NMR (300 MHz, DMSO-d6): δ 8.31 (br t, J = 6.0, 2 H), 7.80-7.73 (m, 6 H), 7.46-7.43 (m, 2 H), 7.34-7.33 (m, 2 H), 7.21-7.17 (m, 2 H), 5.49 (br, 1 H), 4.30-4.18 (m, 5 H), 3.79 (q, J = 6.0, 1 H), 3.69 (d, J = 6.0, 4 H), 1.42 (d, J = 6.0, 6 H), 1.34 (s, 18 H).
2,2'-(((2S,2'S)-2,2'-(((2-Hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2 diyl))bis(propanoyl))bis(azanediyl))diacetic acid (CNU 017). A suspension of compound CNU 016 (70 mg, 0.1 mmol) in THF and 0.1 M sodium hydroxide (2.4 mL) was stirred at 0˚C for 16h. The solution was acidified with 10% HCl and volatiles were removed. The residue was extracted with EtOAc, washed with water and brine, and dried with Na2SO4. After filtration, evaporation and purification of the residue by column chromatography (silica gel, Hexane / EtOAc = 50%, Acetic acid 50 μL), compound CNU 017 (46.2 mg, 78.3%) was finally obtained as a white solid. 1H-NMR (300 MHz, DMSO-d6): δ 12.55 (br, 2 H), 8.29 (br t, J = 6.0, 2 H), 7.80-7.72 (m, 6 H), 7.46-7.43 (m, 2 H), 7.34-7.33 (m, 2 H), 7.21-7.17 (m, 2 H), 5.50 (br d, J = 3.0, 1 H), 4.30-4.15 (m, 5 H), 3.81 (q, J = 7.0, 2 H, overlap with br t), 3.73 (br t, J = 6.0, 4 H, overlap with q), 1.43 (d, J = 9.0, 6 H).
2,2'-(((2-Hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))bis(N-hexylpropanamide) (CNU 018). Following the procedure used to prepare CNU 015, using 2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))dipropionic acid (2, 80 mg, 0.2 mmol), HATU (172.24 mg, 0.45 mmol), n-hexylamine (45.85 mg, 0.45 mmol) and triethylamine (83.82 μL, 0.60 mmol) and eluting with a 20% EtOAc in hexane provided CNU 018 (77.3 mg, 69.3% yield) as an oil. 1H-NMR (300 MHz, CDCl3): δ 7.24-7.20 (m, 4 H), 6.93-6.89 (m, 4 H), 5.26 (br, 2 H), 4.38 (q, J = 6.0, 1 H), 4.16-4.13 (m, 4 H), 3.49 (q, J = 7.0, 2 H). 3.17 (q, J = 6.0, 4 H), 2.56 (br d, J = 6.0, 1 H),1.50 (d, J = 9.0, 6 H), 1.41-1.19 (m, 16 H), 0.85 (t, J = 6.0, 6 H).
Di-tert-butyl 2,2'-((2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))bis(propanoyl))bis(azanediyl))diacetate (CNU 019). Following the procedure used to prepare CNU 015, using 2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))dipropionic acid (2, 100 mg, 0.26 mmol), HATU (125.21 mg, 0.57 mmol), glycine 1,1-dimethylethyl ester (tert-Butyl 2-aminoacetate) (74.30 mg, 0.57 mmol) and triethylamine (107.17 μL, 0.77 mmol) and eluting with a 20% EtOAc in hexane provided CNU 019 (55 mg, 34.8% yield) as an oil. 1H-NMR (300 MHz, DMSO-d6): δ 7.26-7.23 (m, 4 H, overlap with s), 6.93-6.90 (m, 4 H), 5.85 (br, 2 H), 4.34 (q, J = 6.0, 1 H), 4.15-4.13 (m, 4 H), 3.96-3.75 (m, 4 H), 3.56 (q, J = 6.0, 2 H), 1.51 (d, J = 6.0, 6 H), 1.43 (s, 18 H).
2,2'-((2,2'-(((2-Hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))bis(propanoyl))bis(azanediyl))diacetic acid (CNU 020). Following the procedure used to prepare CNU 017, using CNU 019 (120 mg, 0.2 mmol) in THF, 0.1 M sodium hydroxide (4 mL) eluting with a 20% EtOAc in hexane provided CNU 020 (44.2 mg, 45.2% yield) as an oil. 1H-NMR (300 MHz, CDCl3 with DNSO-d6): δ 7.26-7.23 (m, 4 H), 6.91-6.88 (m, 4 H), 6.61 (br, 2 H), 4.35-4.28 (m, 1 H), 4.11 (t, J = 6.0, 4 H), 4.00-3.82 (m, 4 H), 3.60 (q, J = 7.0, 2 H), 1.47 (d, J = 6.0, 6 H).
Dimethyl 2,2'-(((2S,2'S)-2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2-diyl))bis(propanoyl))bis(azanediyl))(2R,2'R)-dipropionate (CNU 021). Following the procedure used to prepare CNU 015, using (2S,2'S)-2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(naphthalene-6,2-diyl))dipropionic acid (1, 100 mg, 0.2 mmol), HATU (228.1 mg, 0.59 mmol), D-alanine methyl ester hydrochloride (83.7 mg, 0.59 mmol) and triethylamine (83.4 μL, 0.59 mmol) and eluting with a 20% EtOAc in hexane provided CNU 021 as a white solid (96.9 mg, 72%). 1H-NMR (400 MHz, DMSO-d6): δ 8.46-8.43 (m, 2 H), 7.80-7.71 (m, 6 H), 7.45-7.42 (m, 2 H), 7.34 (br s, 2 H), 7.21-7.18 (m, 2 H), 5.50 (br d, J = 4.0, 1 H), 4.33-4.16 (m, 7 H), 3.80 (q, J = 8.0, 2 H), 3.64 (s, 3 H), 3.52 (s, 3 H), 1.41 (d, J = 4.0, 3 H), 1.40 (d, J = 4.0, 3 H), 1.28 (d, J = 8.0, 3 H), 1.23 (d, J = 8.0, 3 H).
Dimethyl 2,2'-((2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))bis(propanoyl))bis(azanediyl))(2R,2'R)-dipropionate (CNU 024). Following the procedure used to prepare CNU 015, using 2,2'-(((2-hydroxypropane-1,3-diyl)bis(oxy))bis(4,1-phenylene))dipropionic acid (2, 100 mg, 0.26 mmol), HATU (293.2 mg, 0.77 mmol), D-alanine methyl ester hydrochloride (42.4 mg, 0.31 mmol) and triethylamine (107.2 μL, 0.77 mmol) and eluting with a 20% EtOAc in hexane provided CNU 024 as a white solid (127.1 mg, 87.3%). 1H-NMR (400 MHz, CDCl3): δ 7.24-7.22 (m, 4 H), 6.94-6.90 (m, 4 H), 5.93-5.88 (br m, 2 H), 4.55 (sex, J = 6.7, 2 H), 4.38 (quin, J = 6.0, 1 H), 4.15 (t, J = 6.0, 4 H), 3.72 (s, 3 H), 3.69 (s, 3 H), 3.54 (q, J = 6.7, 2 H), 2.65 (br d, J = 4.0, 1 H), 1.50-1.48 (m, 6 H), 1.34 (d, J = 8.0, 3 H), 1.29 (d, J = 8.0, 3 H).
3. Cell culture
BV2 mouse microglial cells (ATCC, Manassas, VA, USA) were grown in culture media containing Dulbecco’s modified Eagle’s medium (DMEM) (Welgene, Daegu, Korea) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 1% penicillin/streptomycin (Welgene) at 37ºC in a humidified atmosphere of 5% CO2.
4. Preparation of whole-cell lysates
After material treatments with the designated concentrations or time point, BV2 cells were washed with PBS and lysed in a modified radio-immunoprecipitated assay buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.25% sodium deoxycholate, 1% Triton X-100, 1% Nonidet P-40, 1 mM EDTA, 1 mM EGTA, proteinase inhibitor cocktail [1x]). The whole-cell lysates were collected and centrifuged at 14,000 rpm for 15 min at 4°C. The supernatant was preserved, and its protein concentration was determined with Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA).
5. Western blot analysis
An aliquot of proteins (50 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) and transferred onto nitrocellulose membranes (Millipore Co., Bedford, MA, USA). The membranes were washed with Tris-buffered saline (TBS) (10 mM Tris, 150 mM NaCl) supplemented with 0.05% (v/v) Tween-20 (TBST) and blocked with the blocking buffer (TBST containing 5% (w/v) non-fat dried milk). The membranes were incubated overnight with respective primary antibody of p-ERK-1/2 (1:2,000), T-ERK-1/2 (1:2,000), p-JNK-1/2 (1:2,000), T-JNK-1/2 (1:2,000), p-p38 MAPK (1:2,000), T-p38 MAPK (1:2,000), IκB-α (1:2,000), iNOS (1:1,000), or actin (1:10,000) at 4°C. The membranes were washed with TBST and then incubated with secondary antibodies coupled to horseradish peroxidase for 2 h. The membranes were then washed with TBST. Enzyme-linked chemiluminescence (ECL) reagents (Advansta, San Jose, CA, USA) was used to develop the image. Equal loading of proteins was verified by actin antibody.
6. Reverse transcription-polymerase chain reaction (RT-PCR) analysis
After material treatments with the designated concentrations or time point, total RNA from the conditioned BV2 cells was extracted using RNAiso Plus (TaKaRa, Kusatsu, Shiga, Japan). Random hexadeoxynucleotide primer and reverse transcriptase were used for reverse transcribed the total RNA (3 μg). The single-strand cDNA was then amplified by PCR with specific sense and antisense primer of iNOS or actin. The primer sequences used for PCR were as follows: iNOS forward 5’GACAAGCTGCATGTAACATC, and reverse GCTGGTAGGTTCCTGTTGTT3’; actin forward 5’-GGT GAA GGT CGG TGT GAA CG-3’; reverse 5’-GGT AGG AAC ACG GAA GGC CA-3’. Expression levels of actin mRNA were used to evaluate the relative mRNA expression of iNOS.
Results
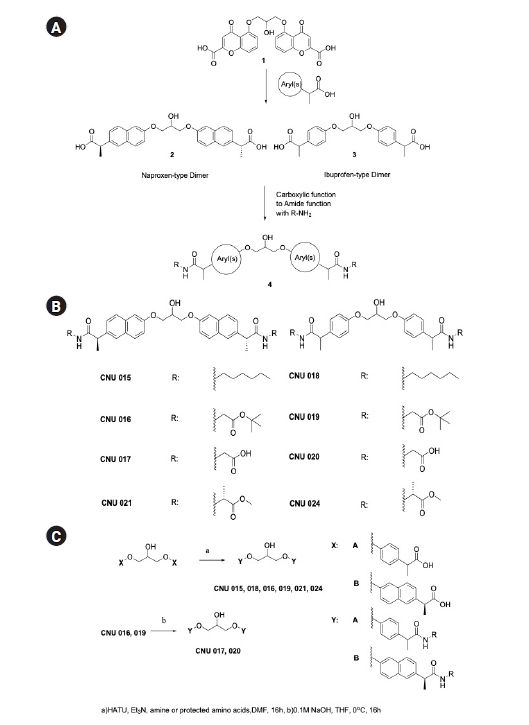
1. Dimer amide design and synthesis
Our synthetic effort to mask carboxylic acid with biologically labile amide bond generated eight new compounds (Fig. 1A-C) in low to moderate yields. For the introduction of amide function, the amino part of protected glycine (CNU 016, 017, 019, and 020) or alanine methyl ester (CNU 021 and 024) were conjugated. In addition, in order to evaluate the structural requirement for the biological activity the carboxylic part of new amide dimers were exposed (CNU 017 and 020). The conjugated derivatives with amines at the position of aryl propionic acid of NSAIDs were reported, but the amide dimers (4, Fig. 1A) were never investigated for the neuroinflammation modulation. The ibuprofen-type and naproxen-type amide dimers were synthesized from carboxylic acid (2, 3, Fig. 1A and 1C) with amine or two equivalents of methyl or t-butyl esters of amide monomer having aryl phenol were reacted with epichlorohydrin under K2CO3 basic conditions to give amide dimers in moderate yield (no experimental and characterization details for CNU 022, 023, 025-027 were provided for the reason of patent and will be reported later).
2. Effects of synthesized amide dimers on the LPS-induced iNOS expression in BV2 cells
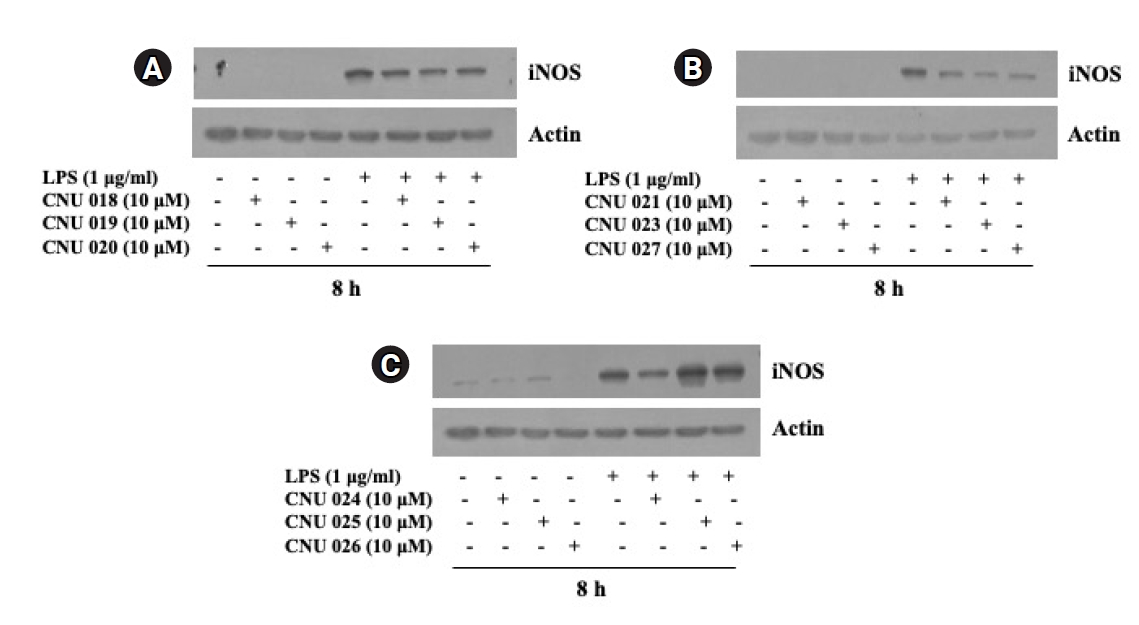
We next investigated the effect of synthesized amide dimers at a concentration of 10 µM on the LPS-induced iNOS expression in BV2 cells for 8 h by using Western blot analysis. As shown in Fig. 2A-C, compared with control (no LPS), there was an elevated iNOS expression in BV2 cells only when exposed to LPS for 8 h. Of note, BV2 cells treated with CNU 019, CNU 020, CNU 021, CNU 023, CNU 024, and CNU 027, respectively, exhibited a substantial reduction in iNOS protein expression levels compared with control. On the contrary, there was no effect on the LPS-induced iNOS expression BV2 cells treated with CNU 025 or 026 compared with control; rather they slightly enhanced it. Total protein expression levels of actin, used and included as an internal control herein, remained constant under these experimental conditions. Due to the strong inhibitory effect on the LPS-induced iNOS expression in BV2 cells, we selected CNU 024 for further studies (No experimental details and results for CNU 015-017 were provided for the reason of different cell lines used and negative results).
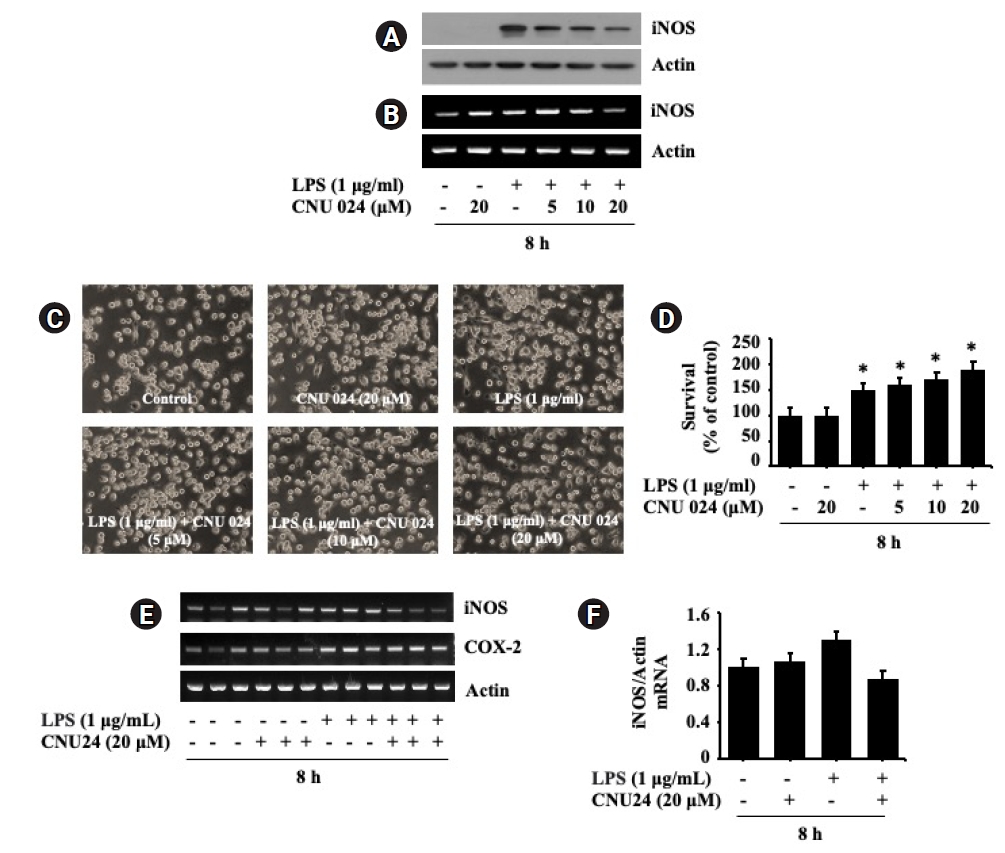
3. CNU 024 at 20 μM has a strong inhibitory effect on the LPS-induced iNOS expression in BV2 cells
We next tested the effect of CNU 024 at different concentrations (5, 10, or 20 μM) on the LPS-induced iNOS protein expression in BV2 cells for 8 h. As shown in Fig. 3A, treatment with CNU 024 resulted in a concentration-dependent inhibition of the LPS-induced iNOS expression in BV2 cells in which maximal inhibition was seen at 20 μM. Total protein expression levels of actin remained unchanged under these experimental conditions. To see whether the CNU 024’s inhibition of the LPS-induced iNOS protein expression herein was due to iNOS transcriptional down-regulation, we next measured the effect of CNU 024 at 5, 10, or 20 μM on the LPS-induced iNOS expression at the mRNA levels in BV2 cells for 8 h by using RT-PCR analysis. As shown in Fig. 3B, treatment with control (no LPS), LPS induced an elevation of iNOS transcripts in BV2 cells. Distinctly, while CNU 024 treatment at 5 or 10 μM had no inhibitory effect on the LPS-induced iNOS mRNA expression in BV2 cells, treatment with CNU 024 at 20 μM markedly repressed the agonist-induced iNOS mRNA expression in these cells. Total mRNA expression levels of actin remained constant under these experimental conditions. These results collectively demonstrate the ability of CNU 024 at 20 μM to strongly inhibit the LPS-induced iNOS expression in BV2 cells. The effect of CNU 024 on microglial cells morphology was assessed next. Primary cultured microglia were exposed to 1 μg/mL LPS in the absence or presence of CNU 024 in different concentrations. As expected, most cells treated with vehicle had a ramified shape, which is the typical morphology of resting microglia (Fig. 3C). In addition, the morphology of cells treated with or without CNU 024 and LPS also did not show any difference in shape of the microglia. Of note, as shown in Figure 3D, CNU 024 increased these cells’ survival by approximately 80% in a dose-dependent manner. Triplicate experiments confirmed the ability of CNU 024 at 20 μM to significantly reduce the mRNA expression levels of iNOS, but not COX-2 in BV2 cells treated by LPS (Fig. 3E). The densitometry results of Fig. 3E for iNOS expression levels standardized to those of control actin is shown in Fig. 3F.
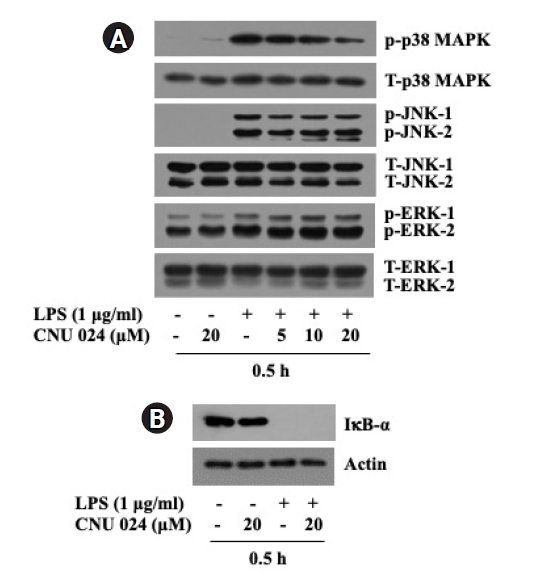
4. CNU 024 at 20 μM exhibits a strong suppressive effect on the LPS-induced p38 MAPK phosphorylation in BV2 cells
We next probed whether CNU 024 modulates the LPS-induced activation of three members of the MAPK family (ERK-1/2, p38 MAPK, and JNK-1/2) and NF-κB, which are reported to be crucial for the agonist-induced iNOS expression in BV2 cells [22,23]. In this study, the activation or inhibition of NF-κB in BV2 cells in response to LPS and/or CNU 024 was assessed by measuring expression levels of IκB-α, an inhibitory cytosolic protein of NF-κB [24,25]. As shown in Fig. 4A, compared with control (no LPS), treatment with LPS at 1 μg/ml for 0.5 h led to a strong phosphorylation of ERK-1/2, p38 MAPK, and JNK-1/2 without influencing respective total protein expression in BV2 cells, pointing out the LPS-induced activation of these MAPKs. As shown in Fig. 4B, the same treatment with LPS also caused a complete loss of IκB-α in BV2 cells compared with control, supporting the agonist-induced activation of NF-κB. Distinctly, while treatment with CNU 024 at doses tested did not largely affect the LPS-induced phosphorylation of ERK-1/2 and JNK-1/2 in BV2 cells, it resulted in a dose-dependent inhibition of the LPS-induced phosphorylation of p38 MAPK in these cells. Total expression levels of ERK-1/2, p38 MAPK, and JNK-1/2 remained unchanged under these experimental conditions. CNU 024 treatment at 20 μM also had no effect on the LPS-induced loss of IκB-α in BV2 cells. These results collectively demonstrate the ability of CNU 024 at 20 μM to strongly and selectively inhibit the LPS-induced p38 MAPK activation in BV2 cells.
Discussion
Microglia activation results in an abnormal elevation of many inflammatory mediators including iNOS and it has been linked to the pathogenesis of neuroinflammation-related neurological disorders. The present study is designed to test a small series of naproxen and ibuprofen amide dimers and evaluate whether these amide dimers have anti-neuroinflammatory effects on the LPS-induced iNOS expression in BV2 mouse microglial cells, a well-established in vitro model of neuroinflammation. Here we show that CNU 024, one of NSAID amide dimers, strongly inhibits the LPS-induced iNOS expression at the protein and mRNA levels in BV2 cells. Our data further provide that the CNU 024’s inhibitory effect on the LPS-induced iNOS expression in BV2 cells is partly due to the inactivation of p38 MAPK pathway.
It has been reported that the disodium salt of cromoglicic acid (1, Fig. 1A) used mainly for asthma [26] has the anti-Aꞵ aggregation property in vitro and reduces the AD-associated Aꞵ protein expression levels by increasing microglial phagocytosis [27,28]. NSAIDs, such as ibuprofen and naproxen, that are used to treat pain, fever, and inflammation, are shown to possess anti-aggregation properties of Aꞵ proteins and anti-neuroinflammatory activities [29-32]. Notably, our previous studies have shown that the replacement of chromone rings in cromoglicic acid (1, Fig. 1A) with 2-aryl propionic acids as a part of pharmacophore in NSAIDs provides dimer structure (2, naproxen-type and 3, ibuprofen-type dimers, Fig. 1A) with anti-neuroinflammatory function and Aꞵ aggregation inhibition (data not shown). However, the presence of carboxylic acid moiety still can cause the gastrointestinal side effects as shown by NSAIDs having 2-aryl(s) propionic acid moiety and low brain uptake [33,34]. One of the approaches to address this problem could be masking carboxylic function with appropriate molecules as in a form of amide linkage while maintaining anti-neuroinflammatory function (4, Fig. 1A). Pharmacokinetic studies of naproxen amides have pointed out some amino acid esters with promising colorectal cancer chemopreventive activity [35]. Therefore, in this study, we investigated whether the structural modification with amide function of two NSAIDs naproxen and ibuprofen still hold the ability to modulate neuroinflammation, and to evaluate the necessity of carboxylic function as a part of our ongoing effort to generate multi-target ligands for AD [36].
In accordance with this understanding, we screened 8 compounds derived from 2-aryl propionic acid amide modification of naproxen and ibuprofen dimers and conveyed that among them, CNU 019, 020, 021, 023, 024, and 027 at 10 µM for 8 h, showed anti-inflammatory effect by substantially inhibiting iNOS expression induced by LPS in BV2 cells (Fig. 2A-C). Through additional dose-response experiments, we further confirmed the ability of CNU 024 mainly at 20 µM to vastly inhibit the LPS-induced iNOS expression at the protein and mRNA levels in BV2 cells (Fig. 3A, B). These results strongly point out that CNU 024 exerts its inhibitory effect on the LPS-induced iNOS expression through the iNOS transcriptional downregulation in BV2 cells.
It is known that neuroinflammation is increased in the damaged brain, which could be found in many neurological disorders, and it can be induced by the activation of NF-κB [24,25]. NF-κB is a family of structurally related transcription factors that is involved in the control of various inflammatory and immune responses, cell growth and apoptosis [37]. Several inducers including LPS have been known as NF-κB activator [37]. Accordingly, the LPS-induced NF-κB activation is mainly mediated though rapid proteolysis (protein degradation) of IκB-α, the NF-κB inhibitory cytosolic protein [24], which is crucial for the agonist-induced iNOS expression in BV2 cells [38]. In agreement with it, here, we demonstrated the activation of NF-κB in BV2 cells exposed to LPS for 0.5 h, as evidenced by the agonist’s ability to completely deplete the expression levels of IκB-α (Fig. 4B). Up to date, CNU 024’s regulation of the LPS-induced NF-κB activation (proteolysis of IκB-α) in BV2 cells is not reported. The present study, however, showed that the LPS-induced proteolysis of IκB-α in BV2 cells is not influence by CNU 024. Reportedly, the LPS-induced iNOS expression in BV2 cells are associated with not only NF-κB but also other transcriptional factors [22]. It is thus likely that the CNU 024’s inhibitory effect on the LPS-induced iNOS expression in BV2 cells is mediated not through regulation of NF-κB/IκB-α but via that of other transcriptional factors and signaling pathways or components.
Studies have previously shown that activation of the MAPK family, including ERK-1/2, JNK-1/2, and p38 MAPK, are important for the LPS-induced iNOS expression in BV2 cells [39,40]. In the current study, treatment with LPS for 0.5 h leads to the phosphorylation (activation) of p38 MAPK, ERK-1/2, and JNK-1/2 in BV2 cells (Fig. 4A), supporting the LPS’s stimulating effects on these signaling components. Of note, we observed that CNU 024 at 20 μM dramatically lowers the LPS-induced phosphorylated levels of p38 MAPK in BV2 cells, whereas it does not influence the agonist-induced phosphorylated levels of ERK-1/2 and JNK-1/2 in these cells. These results strongly indicate that CNU 024 exerts its inhibitory effect on the LPS-induced iNOS expression in BV2 cells not through regulation of the ERK-1/2 and JNK-1/2 but via control (inhibition) of the p38 MAPK and its downstream signaling pathway.
Conclusions
It is the first study to demonstrate that CNU 024, one of the NSAID amide dimers, strongly suppresses the induction of iNOS expression in LPS-stimulated BV2 murine microglial cells, and the suppression is partly due to the selective inhibition of p38 MAPK. This work shows that CNU 024 could be a valuable ligand for further development as a potential drug candidate for treating neuroinflammatory pathologies.